XTRACT - a command line tool for automated tractography
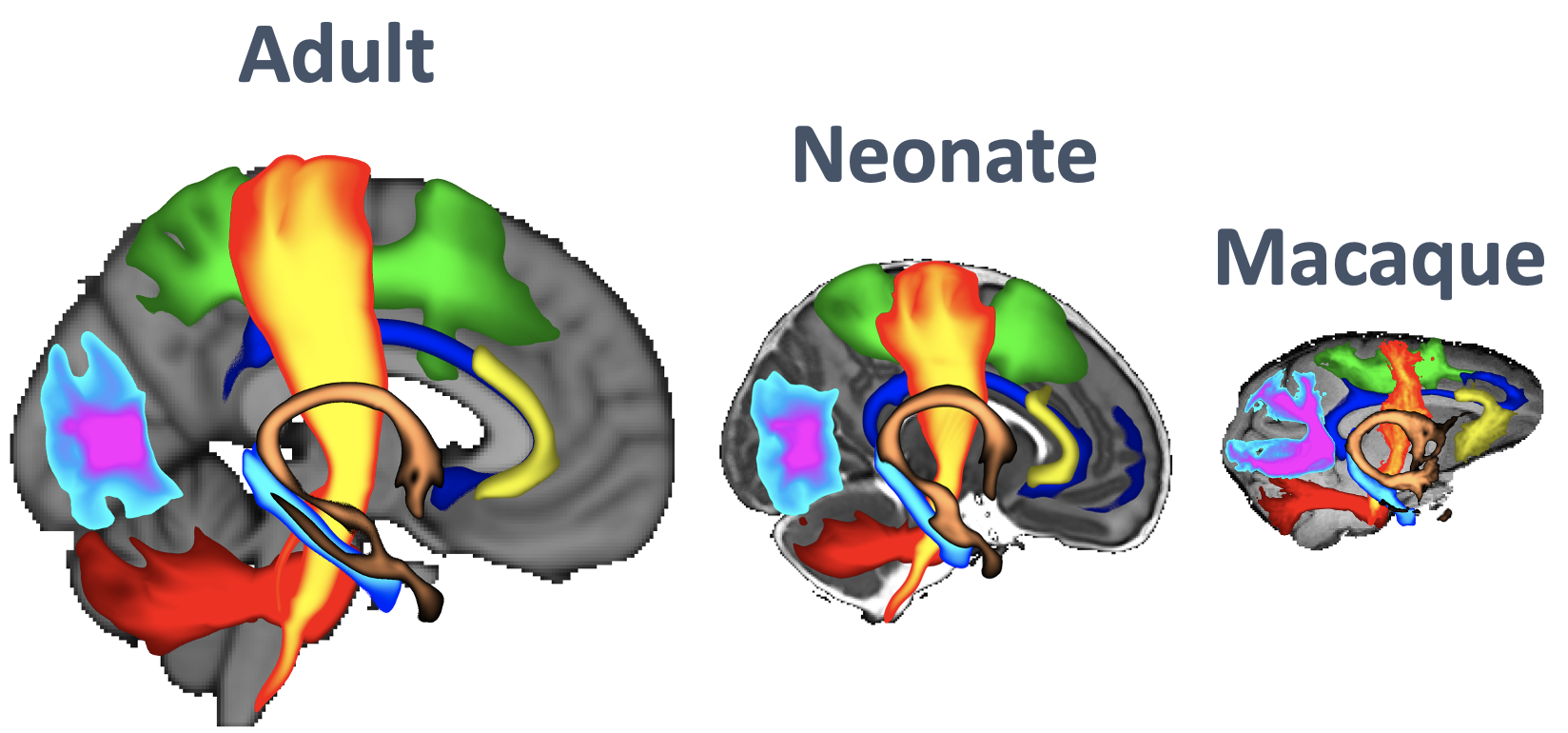
XTRACT (cross-species tractography) can be used to automatically extract a set of carefully dissected tracts in human (neonates and adults) and macaques. It can also be used to define one's own tractography protocols where all the user needs to do is to define a set of masks in standard space (e.g. MNI152).
XTRACT reads the standard space protocols and performs probabilistic tractography (probtrackx in the subject's native space. Resultant tracts may be stored in either the subject's native space or in standard space. The user must provide the crossing fibres fitted data (bedpostx) and diffusion to standard space registration warp fields (and their inverse).
XTRACT atlases are available within FSLeyes or can be downloaded here.
This page is organised into the following sections:
Citation
If you use XTRACT in your research, please cite the following articles:
Warrington S., Bryant K., Khrapitchev A., Sallet J., Charquero-Ballester M., Douaud G., Jbabdi S.*, Mars R.*, Sotiropoulos S.N.* (2020) XTRACT - Standardised protocols for automated tractography and connectivity blueprints in the human and macaque brain. NeuroImage, 217(116923). DOI: 10.1016/j.neuroimage.2020.116923
Warrington S.*, Thompson E.*, Bastiani M., Dubois J., Baxter L., Slater R., Jbabdi S., Mars R.B., Sotiropoulos S.N. (2022) Concurrent mapping of brain ontogeny and phylogeny within a common space: Standardzed tractography and applications. Science Advances, 8(42). DOI: 10.1126/sciadv.abq2022
de Groot M., Vernooij M.W. Klein S., Ikram M.A., Vos F.M., Smith S.M., Niessen W.J., Andersson J.L.R. (2013) Improving alignment in Tract-based spatial statistics: Evaluation and optimization of image registration. NeuroImage, 76(1), 400-411. DOI: 10.1016/j.neuroimage.2013.03.015
If you use the macaque protocols, please cite:
Assimopoulos, S., Warrington, S., Bryant, K.L., Pszczolkowski, S., Jbabdi S., Mars R., Sotiropoulos S.N. Generalising XTRACT tractography protocols across common macaque brain templates. Brain Struct Funct (2024). DOI: 10.1007/s00429-024-02760-0
XTRACT
XTRACT is a command-line tool. XTRACT automatically detects if $SGE_ROOT is set and if so uses fsl_sub. For optimal performance, use the GPU version!
Simply type xtract or xtract --help to get the usage:
__ _______ ____ _ ____ _____
\ \/ /_ _| _ \ / \ / ___|_ _|
\ / | | | |_) | / _ \| | | |
/ \ | | | _ < / ___ \ |___ | |
/_/\_\ |_| |_| \_\/_/ \_\____| |_|
usage: XTRACT [-h] [-bpx <folder>] [-out <folder>] [-species <SPECIES>]
[-tract_list <list>] [-str <file>] [-p <folder>]
[-stdref <reference>] [-stdwarp <path> <path>] [-native]
[-ref <path> <path> <path>] [-res <mm>] [-ptx_options <file>]
[-interp <str>] [-gpu] [-par] [-print_list] [-version]
XTRACT: Cross-species tractography
options:
-h, --help show this help message and exit
Required arguments:
-bpx <folder> Path to bedpostx folder
-out <folder> Path to output folder
-species <SPECIES> One of HUMAN or MACAQUE or HUMAN_BABY or CUSTOM.
Optional arguments:
-tract_list <list> Comma separated tract list defining the subset of
tracts to run (must follow xtract naming convention)
-str <file> Structures file (format: <tractName> [samples=1], 1
means 1000, '#' to skip lines)
-p <folder> Protocols folder (all masks in same standard space)
(Default=/home/paulmc/fsl/fsl-
latest/data/xtract_data/<SPECIES>)
-stdref <reference> Standard space reference image (Default =
/home/paulmc/fsl/fsl-
latest/data/standard/MNI152_T1_1mm [HUMAN],
/home/paulmc/fsl/fsl-
latest/data/xtract_data/standard/F99/mri/struct_brain
[MACAQUE], /home/paulmc/fsl/fsl-latest/data/xtract_dat
a/standard/neonate/mri/schuh_template [HUMAN_BABY]
-stdwarp <path> <path>
<std2diff> <diff2std> Non-linear Standard2diff and
Diff2standard transforms (Default=bedpostx_dir/xfms/{s
tandard2diff_warp.nii.gz,diff2standard_warp.nii.gz})
-native Run tractography in native (diffusion) space
-ref <path> <path> <path>
<refimage> <diff2ref> <ref2diff> Reference image for
running tractography in reference space,
Diff2Reference and Reference2Diff transforms
-res <mm> Output resolution (Default=same as in protocol folders
unless '-native' used)
-ptx_options <file> Pass extra probtrackx2 options as a text file to
override defaults, e.g. --steplength=0.2
--distthresh=10)
-interp <str> If native/ref: default interpolation of protocol ROIs
is nearest neighbour ('nn', since vXXX). For backwards
compatability, trilinear plus thresholding ('tri') is
available
-gpu Use GPU version
-par If cluster, run in parallel: submit 1 job per tract
-print_list List the tract names used in XTRACT
-version List the package versions
Example usage:
xtract -bpx /data/Diffusion.bedpostX -out /data/xtract -species HUMAN -stdwarp std2diff.nii.gz diff2std.nii.gz -gpu
Outputs of XTRACT
Under <outputDir>:
commands.txt- XTRACT processing commandslogs- directory containing the probtrackx log filestracts- directory containing tractography results-
<tractName>- directory per tract, each containing:waytotal- text file containing the number of valid streamlinesdensity.nii.gz- nifti file containing the fibre probability distributiondensity_lenths.nii.gz- nifti file containing the fibre lengths, i.e. each voxel is the average streamline length - this is the-omplprobtrackx optiondensityNorm.nii.gz- nifti file containing the waytotal normalised fibre probability distribution (the -density.nii.gzdivided by the total number of valid streamlines)- if the protocol calls for reverse-seeding:
tractsInv- directory containing the above for the seed-target reversed runsum_waytotalandsum_density.nii.gz- the summed waytotal and fibre probability distribution
-
If the -native option is being used:
- masks - directory
- <tractName> - directory per tract containing the native space protocol masks
Note
The primary output is the densityNorm.nii.gz file.
Pre-processing
Prior to running XTRACT, you must complete the following steps from FDT processing pipeline:
- Brain extraction using BET
- Susceptibility distortion correction using topup (only if spin-echo fieldmaps have been acquired - if you don't have these, skip to step 3)
- Eddy current distortion and motion correction using eddy
- Fit the crossing fibre model using bedpostx
- Non-linear registration to standard space (MNI152 for adult human, F99 for macaque), see the FDT pipeline
Your data should now be ready to run XTRACT!
Note on ANTs warp fields
To use these fields, you must convert the warp fields to FSL's FNIRT convention. We provide a script to do so here.
Standard Spaces
- For HUMAN, XTRACT uses the MNI152 standard space in
$FSLDIR/etc/standard - For MACAQUE, XTRACT uses the F99 atlas in Caret - see http://brainvis.wustl.edu/wiki/index.php/Caret:Atlases
We also provide a copy of the F99 atlas in
$FSLDIR/etc/xtract_data/standard/F99. This includes a helper script for registering your own diffusion/structural data to the F99 atlas.- MACAQUE protocols are now available in five commonly used templates spaces. Protocols and templates can be found under
$FSLDIR/etc/xtract_data/. See 10.1007/s00429-024-02760-0 for full details.
- MACAQUE protocols are now available in five commonly used templates spaces. Protocols and templates can be found under
- For BABY, XTRACT uses the Schuh 40-week PMA template - see https://doi.org/10.1101/251512
XTRACT Tracts
Automatically Extracted Tracts
When running XTRACT with the -species option, a predefined list of tracts is automatically extracted. Currently the following tracts are available (all split into left/right except the commissural tracts):
| Tract | Abbreviation |
|---|---|
| Arcuate Fasciculus | AF |
| Acoustic Radiation | AR |
| Anterior Thalamic Radiation | ATR |
| Cingulum subsection : Dorsal | CBD |
| Cingulum subsection : Peri-genual | CBP |
| Cingulum subsection : Temporal | CBT |
| Corticospinal Tract | CST |
| Frontal Aslant | FA |
| Forceps Major | FMA |
| Forceps Minor | FMI |
| Fornix | FX |
| Inferior Longitudinal Fasciculus | ILF |
| Inferior Fronto-Occipital Fasciculus | IFO |
| Middle Cerebellar Peduncle | MCP |
| Middle Longitudinal Fasciculus | MdLF |
| Optic Radiation | OR |
| Superior Thalamic Radiation | STR |
| Superior Longitudinal Fasciculus 1 | SLF1 |
| Superior Longitudinal Fasciculus 2 | SLF2 |
| Superior Longitudinal Fasciculus 3 | SLF3 |
| Anterior Commissure | AC |
| Uncinate Fasciculus | UF |
| Vertical Occipital Fasciculus | VOF |
You may specify a subset of these tracts using the -tract_list option.
Adding your own tracts
Suppose you want to create an automated protocol for a tract called mytrack.
First you need to create a folder called mytrack which you can add e.g. in the protocols folder.
Then create the following NIFTI files (with this exact naming) and copy them into mytrack:
Compulsory:
- seed.nii.gz : a seed mask
Optional:
- stop.nii.gz : a stop mask if required
- exclude.nii.gz : an exclusion mask if required
- ONE of the following:
- target.nii.gz : a single target mask
- target1.nii.gz, target2.nii.gz, etc. : a number of targets, in which case streamlines will be kept if they cross ALL of them
- invert (empty file to indicate that a seed->target and target->seed run will be added and combined) \
if such an option is required, a single target.nii.gz file is also expected
All the masks above should be in standard space (e.g. MNI152 or F99) if you want to run the same tracking for a collection of subjects.
Next, make a structure file using the format <tractName> <nsamples>per line and call XTRACT using -species <SPECIES> -str <file> -p <folder>, pointing to your new protocols folder 'mytrack'.
External Compatible Protocol Libraries
Additional tractography protocols, compatible with XTRACT, are available for other species and brains.
- Chimpanzee: Bryant et al. (2020) A comprehensive atlas of white matter tracts in the chimpanzee. PLoS Biol 18(12): e3000971
- Pig: Benn et al. (2020) A Pig White Matter Atlas and Common Connectivity Space Provide a Roadmap for the Introduction of a New Animal Model in Translational Neuroscience bioRxiv
- 8 non-human primate species: Bryant et al. (2021) Diffusion MRI data, sulcal anatomy, and tractography for eight species from the Primate Brain Bank. Brain Structure and Function
- Macaque, Gorilla, Chimpanzee, Human: Roumazeilles et al. (2020) Longitudinal connections and the organization of the temporal cortex in macaques, great apes, and humans. PLOS Biology
- Lar gibbon: Bryant et al. (2023) A map of white matter tracts in a lesser ape, the lar gibbon. Brain Structure and Function
If you have made XTRACT-style protocols and want them to be listed here, get in touch!
To implement external libraries:
- download the protocols and standard space from the relevant publication,
- build a structure list (
<tractName> <nsamples>per line), - follow the pre-processing steps described above to obtain warp fields between the native diffusion space and the relevant standard space (the space in which the protocols are defined),
- call XTRACT using the
-species CUSTOMargument, specifying the standard space reference, the protocols directory and the structure list file (available in FSL 6.0.5+)
Running XTRACT with other species:
After developing your own protocols for a given species, you may use the -species CUSTOM argument to run XTRACT on any species.
To do so you must provide a standard space brain in addition to specifying the protocol folder and structure file.
XTRACT Atlases
42 probabilistic tract atlases were derived using 1021 subjects from the Human Connectome Project as described in Warrington et al. (2020) NeuroImage. In brief, XTRACT was applied to each subject’s minimally preprocessed diffusion MRI data (Glasser et al, Neuroimage 2013 - Sotiropoulos et al, Neuroimage 2013). The subsequent tract estimates were binarised at a threshold and averaged across the cohort, providing population percent overlap maps for each of the major white matter fibre bundles.
Data were provided by the Human Connectome Project, WU-Minn Consortium (Principal Investigators: David Van Essen and Kamil Ugurbil; 1U54MH091657) funded by the 16 NIH Institutes and Centers that support the NIH Blueprint for Neuroscience Research; and by the McDonnell Center for Systems Neuroscience at Washington University.
We also include equivalent tract atlases for the macaque brain.
These atlases are available within FSLeyes or can be downloaded here.
Citation: Warrington S, Bryant K, Khrapitchev A, Sallet J, Charquero-Ballester M, Douaud G, Jbabdi S, Mars R, Sotiropoulos SN* (2020) XTRACT - Standardised protocols for automated tractography and connectivity blueprints in the human and macaque brain. NeuroImage. DOI: 10.1016/j.neuroimage.2020.116923
XTRACT blueprint
Generating Connectivity Blueprints with xtract_blueprint
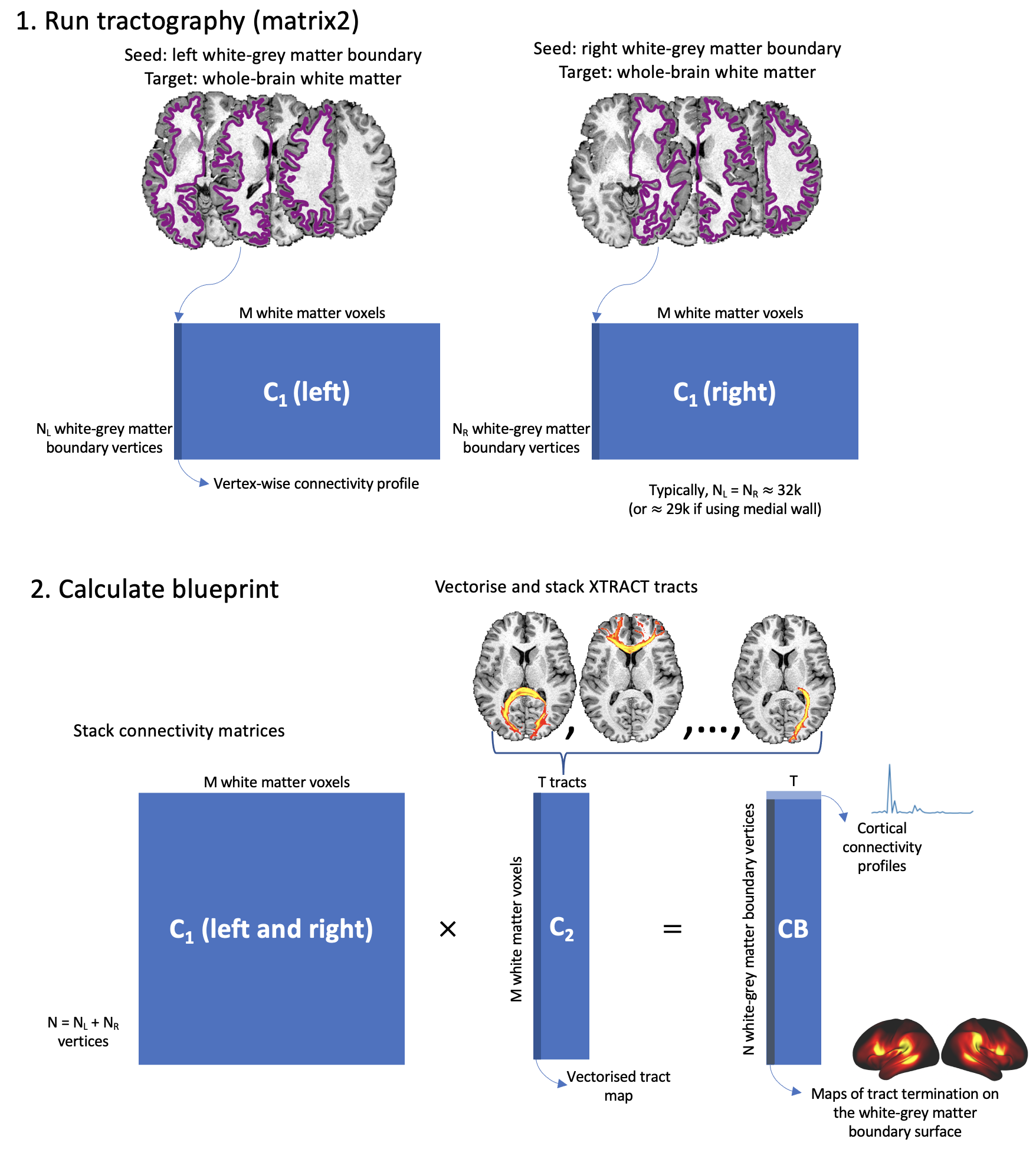
xtract_blueprint is a flexible yet simple way to calculate the connectivity blueprint (Mars et al. 2018 eLife, Warrington et al. 2020 NeuroImage, Warrington et al. 2022 SciAdv): a vertex (or voxel) by tract matrix where each row describes how each (sub)cortical location is connected to major white matter fibre bundles, and each column describes the (sub)cortical terminations of each of the white matter fibre bundles. xtract_blueprint can, in principle, build a connectivity blueprint for any brain (e.g. different species), at any resolution (i.e. number of vertices) and for any region (whole whole or a given ROI, a lobe for example), including the subcortex.
The script was written by Shaun Warrington (University of Nottingham), Saad Jbabdi (Oxford University) and Stamatios Sotiropoulos (University of Nottingham).
The below figure summarises the steps that xtract_blueprint runs to obtain the blueprint matrix.
Citations
Mars R., Sotiropoulos S.N., Passingham R.E., Sallet J., Verhagen L., Khrapitchev A.A., Sibson N., Jbabdi S. (2018) Whole brain comparative anatomy using connectivity blueprints. eLife. DOI: 10.7554/eLife.35237
Warrington S.*, Thompson E.*, Bastiani M., Dubois J., Baxter L., Slater R., Jbabdi S., Mars R.B., Sotiropoulos S.N. (2022) Concurrent mapping of brain ontogeny and phylogeny within a common space: Standardzed tractography and applications. Science Advances, 8(42). DOI: 10.1126/sciadv.abq2022
Warrington S., Bryant K., Khrapitchev A., Sallet J., Charquero-Ballester M., Douaud G., Jbabdi S.*, Mars R.*, Sotiropoulos S.N.* (2020) XTRACT - Standardised protocols for automated tractography in the human and macaque brain. NeuroImage. DOI: 10.1016/j.neuroimage.2020.116923
Usage
__ _______ ____ _ ____ _____ _ _ _ _
\ \/ /_ _| _ \ / \ / ___|_ _| |__ | |_ _ ___ _ __ _ __(_)_ __ | |_
\ / | | | |_) | / _ \| | | | | '_ \| | | | |/ _ \ '_ \| '__| | '_ \| __|
/ \ | | | _ < / ___ \ |___ | | | |_) | | |_| | __/ |_) | | | | | | | |_
/_/\_\ |_| |_| \_\/_/ \_\____| |_| |_.__/|_|\__,_|\___| .__/|_| |_|_| |_|\__|
|_|
usage: XTRACT Blueprint [-h] -bpx <folder> -xtract <folder> -seeds <list>
-target <mask> [-warps <path> <path> <path>] [-native]
[-out <folder>] [-stage {0,1,2}] [-savetxt]
[-prefix <string>] [-rois <list>] [-stops <stop.txt>]
[-wtstops <wtstop.txt>] [-exclusion <nifti>]
[-subseed <subseed.txt>] [-tract_list <list>]
[-thr <float>] [-nsamples <int>] [-res <float>]
[-ptx_options <options.txt>] [-gpu] [-keepfiles]
[-version]
xtract_blueprint: for generating connectivity blueprints from xtract output
options:
-h, --help show this help message and exit
Required arguments:
-bpx <folder> Path to bedpostx folder
-xtract <folder> Path to xtract folder
-seeds <list> Comma separated list of seeds for which a blueprint is
requested (e.g. left and right cortex in standard
space)
-target <mask> A whole brain/WM binary target mask in the same space
as the seeds
Optional arguments:
-warps <ref> <xtract2diff> <diff2xtract> Standard space
reference image, and transforms between xtract space
and diffusion space
-native Run tractography in native (diffusion) space
-out <folder> Path to output folder (default is
<xtract_dir>/blueprint)
-stage {0,1,2} What to run. 0:everythng (default), 1:matrix2,
2:blueprint
-savetxt Save blueprint as txt file (nseed by ntracts) instead
of CIFTI
-prefix <string> Specify a prefix for the final blueprint filename
(e.g. <prefix>_BP.LR.dscalar.nii)
-rois <list> Comma separated list (1 per seed): ROIs (gifti) to
restrict seeding (e.g. medial wall masks)
-stops <stop.txt> Text file containing line separated list of stop masks
(see probtrackx usage for details)
-wtstops <wtstop.txt>
Text file containing line separated list of wtstop
masks (see probtrackx usage for details)
-exclusion <nifti> NIFTI containing a binary exclusion mask
-subseed <subseed.txt>
Text file containing line separated list of
subcortical seeds
-tract_list <list> Comma separated list of tracts to include (default =
all found under -xtract <folder>)
-thr <float> Threshold applied to XTRACT tracts prior to blueprint
calculation (default = 0.001)
-nsamples <int> Number of samples per seed used in tractography
(default = 1000)
-res <float> Resolution of matrix2 output (Default = 3 mm). Set to
0 for standard space resolution.
-ptx_options <options.txt>
Pass extra probtrackx2 options as a text file to
override defaults
-gpu Use GPU version
-keepfiles Do not delete temp files
-version List the package versions
Example usage:
xtract_blueprint -bpx /data/Diffusion.bedpostX -out /data/blueprint -xtract /data/xtract
-seeds l.white.surf.gii,r.white.surf.gii -target WM_segmentation.nii.gz
-rois l.medwall.shape.gii,r.medwall.shape.gii
-warps ref.nii.gz xtract2diff.nii.gz diff2xtract.nii.gz -gpu
Running XTRACT blueprint
In order to use xtract_blueprint, you need to have run xtract first. To construct the connectivity blueprints, xtract_blueprint expects the same warp fields as used in the running of xtract.
xtract_blueprint supports the construction of connectivity blueprints using surface (GIFTI) or volume (NIFTI) files. Surface or volume files may be used for the cortical (white-grey matter boundary) seed. Volume files are required for subcortical seeds. If a volume file is used as the cortical seed, the resultant output will be a 4D NIFTI blueprint. For surface data, the resultant output will be a dscalar CIFTI.
Required input:
- bedpostx folder - crossing fibre modelled diffusion data (expects to find nodif_brain_mask)
- xtract folder - xtract tract folder (the output from xtract)
- seed - the comma separated seed masks to use in tractography (e.g. the white-grey matter boundary surfaces), e.g. L.white.surf.gii, R.white.surf.gii
- warps - a reference standard space image and warps to and from the native diffusion and standard spaces, e.g. MNI152.nii.gz standard2diff.nii.gz diff2standard.nii.gz
- target - a whole-brain white matter target NIFTI mask (resolution of this target is set by the '-res' flag)
Note
If running whole-brain (recommended), you must provide the left seed first, as exampled. We also recommend that you use a medial wall mask to restrict the blueprint to the GM surface.
Running modes: - Stage 1 only - only run seed-based tractography - Stage 2 only - only run blueprint processing (requires xtract output and tractography from stage 1) - All (default) - runs both stage 1 and stage 2 processing
xtract_blueprint is capable of GPU acceleration (-gpu flag) and works with fsl_sub to submit jobs in parallel. If using the CPU version, expect tractography to take many hours - you can speed this up by using a lower resolution seed and/or target.
Tractography details and options:
Tractography is performed for each seed region separately. The resultant connectivity matrices (fdt_matrix) are stacked in order to construct a whole-brain connectivity blueprint. You may also provide a single hemisphere if you wish.
Optionally, you may also provide stop (stop tracking at locations given by this mask file) and wtstop (allow propagation within mask but terminate on exit) masks. Stop is typically the pial surface. wtstop is typically subcortical structures and/or the white surface. These should be specified as line separated text files. e.g. -seeds <l.white.surf.gii,r.white.surf.gii> -stop stop.txt -wtstop wtstop.txt
Spatial resolution: by default tractography will be ran and stored using a resolution of 3 mm for the target. This may be adjusted using the -res argument. Note: if required, xtract_blueprint will resample the xtract tracts. Warning: connectivity matrices are very large and require a lot of memory to handle - 3 mm is usually sufficient for the adult human brain.
Additional probtrackx options may also be supplied. Simply add the probtrackx arguments to a text file and direct xtract_blueprint to this using the -ptx_options argument.
Connectivity blueprints are primarily concerned with the connectivity of the grey matter to white matter tracts. As such, we offer the option the mask out the medial wall. To do so, provide a single medial wall mask per supplied seed: e.g. -seeds l.white.surf.gii,r.white.surf.gii -rois l.roi,r.roi. By default, the medial wall is included in the calculation of the connectivity blueprint: we recommend the use of the medial wall mask to prevent this.
The -roi argument may be used to restrict the blueprint to any region of interest, not just to exclude the medial wall. For example, you may provide an ROI restricting the blueprint to the temporal or frontal lobe.
If you wish to use a stop/wtstop surface mask, you must ensure that the number of vertices matches the seed mask. This means that, if you are providing a seed mask and medial wall mask to xtract_blueprint, and wish to provide a surface stop mask, you must convert the stop mask to asc, restricting the data points to the medial wall mask, e.g.:
${FSLDIR}/bin/surf2surf -i stop.L.surf.gii -o stop.L.asc --outputtype=ASCII --values=l.roi.shape.gii
${FSLDIR}/bin/surf2surf -i stop.R.surf.gii -o stop.R.asc --outputtype=ASCII --values=r.roi.shape.gii
Then supply stop.L.asc and stop.R.asc in a text file to xtract_blueprint using the -stop argument. This conversion is automatically performed for the seed mask in xtract_blueprint if a medial wall mask is supplied.
Subcortical seeding has now been introduced and may be used with the -subseed flag. This should be a line separated text file which each line provide the absolute path to a NIFIT format subcortical structure (should be a binary mask). For xtract_blueprint to generate "proper" CIFTI files, the subcortical structures should follow the CIFTI naming conventions: e.g. CIFTI_STRUCTURE_THALMUS_LEFT, CIFTI_STRUCTURE_AMYGDALA_LEFT, etc...
Which tracts are included?
Connectivity blueprints may be constructed using the provided XTRACT tracts or using your own. By default, xtract_blueprint will use all tracts it finds under the xtract folder. You can specify a subset, or you own tracts, by providing a comma separated list of tracts using the -tract_list argument.
Certain tracts, e.g. the Middle Cerebellar Peduncle (MCP), do not project to the cortex. As such, they should be disregarded when interpreting the connectivity blueprint, or excluded from its construction.
Only interested in the connectivity of a specific area?
In many cases, the connectivity to a particular lobe, e.g. temporal or frontal, is of interest. You can use xtract_blueprint to obtain a connectivity blueprint for such a region:
- Define a binary mask which contains the region of interest as a
shape.giiorfunc.giifile - Select the tracts of interest: in all likelihood, only a subset of XTRACT's tracts will project to the ROI
- Supply the whole white matter surface file along with the ROI to
xtract_blueprint, e.g. for the temporal lobe
Outputs of XTRACT blueprint
xtract_blueprint will create an output directory specified by the -out argument. This will contain any log and command files along with a sub-directory per seed. Each sub-directory contains the resultant connectivity matrix from stage 1. The connectivity blueprint will be saved in the parent output directory (a CIFTI dscalar.nii file).
Under outputDir:
- Stage 1 (matrix2 tractography) output
- omatrix2:
- ptx_commands.txt - the probtrackx commands for tractography
- <seed> - sub-directory containing tractography results for each seed supplied, each containing:
- coords_fdt_matrix2 - the coordinates of the seed mask
- lookup_tractspace_fdt_matrix.nii.gz - the target lookup space
- tract_space_coords_for_fdt_matrix2 - the coordinates of the target mask
- probtrackx.log - the probtrackx log file
- fdt_paths.nii.gz - a NIFTI file containing the generated streamlines
- fdt_matrix.dot - the sparse format connectivity matrix (used to calculate the blueprint)
- waytotal - txt file containing the number of valid streamlines
- Stage 2 (blueprint calculation) output
- bp_commands.txt - the blueprint calculation commands
- BP.<L,R,LR>.dscalar.nii - CIFTI file containing the whole-brain connectivity blueprint
- logs - sub-directory containing job scheduler log files for both stages
Alternatively, the -savetxt option may be used to override this. In this case, two txt files will be saved: the first (BP.<L,R,LR>.txt) will be an n_seed by n_tracts array containing the blueprint; the second (tract_order.<L,R,LR>.txt) is an n_tracts by 1 array containing the tract order in which the blueprint is structured. Note: no CIFTI file will be generated. If a volume seed is provided, xtract_blueprint will generate a volume-space blueprint as a 4D NIFTI file.
XTRACT viewer
Visualising results in FSLeyes with
xtract_viewer
The output of XTRACT is a folder that contains tracts in separate folders. We provide a convenient script (xtract_viewer) that can load these tracts (or a subset of the tracts) into FSLeyes using different colours for the different tracts but matching the left/right colours.
The script was written by Shaun Warrington.
Notice that in the FSL 6.0.2 release, xtract_viewer has not been included into the main $FSLDIR/bin path of executables, therefore it needs to be manually launched from
This has been corrected in FSL 6.0.3+
Usage
__ _______ ____ _ ____ _____ _
\ \/ /_ _| _ \ / \ / ___|_ _| __ _(_) _____ _____ _ __
\ / | | | |_) | / _ \| | | | \ \ / / |/ _ \ \ /\ / / _ \ '__|
/ \ | | | _ < / ___ \ |___ | | \ V /| | __/\ V V / __/ |
/_/\_\ |_| |_| \_\/_/ \_\____| |_| \_/ |_|\___| \_/\_/ \___|_|
usage: XTRACT Viewer [-h] -xtract <folder> [-species <SPECIES>]
[-brain <nifti>] [-tract_list <list>]
[-thr <float> <float>] [-print_cmd]
xtract_viewer: quick visualisation of xtract results in fsleyes
options:
-h, --help show this help message and exit
Required arguments:
-xtract <folder> Path to XTRACT output folder
-species <SPECIES> One of HUMAN or MACAQUE or HUMAN_BABY or CUSTOM. Can
also specify the macaque template space to be used by
appending _[D99,INIA,NMT,YRK] (default is F99).
Optional arguments:
-brain <nifti> Path to custom brain teplate
-tract_list <list> Comma separated list of tracts to include (default =
all found under -xtract <folder>)
-thr <float> <float> Upper and lower threshold applied to tracts for
viewing (default = [0.001 0.1]).
-print_cmd Print the fsleyes command?
Example usage:
xtract_viewer -xtract /data/xtract -species HUMAN
XTRACT stats
Extracting tract-wise summary statistics with xtract_stats
A common usage of the XTRACT output is to summarise tracts in terms of simple summary statistics, such as their volume and microstructural properties (e.g. mean FA). We provide xtract_stats to get such summary statistics in a quick and simple way.
The script was written by Shaun Warrington.
You can use xtract_stats with any modelled diffusion data, e.g. DTI, bedpostx, DKI. It will provide raw and tract probability-weighted statistics.
Simply provide; the directory (and basename of files, if any) leading to the diffusion data of interest, the directory containing the XTRACT output, the warp field (or use 'native' if tracts are already in diffusion space). If tracts are not in diffusion space, you must also provide a reference image in diffusion space (e.g. FA map).
The output (a .csv file) by default contains the tract volume (mm3) and the mean, median and standard deviation of the probability, length, FA and MD for each tract.
e.g. call for crossing fibre features and tract volume:
xtract_stats -d /home/DTI.bedpostX/mean_ -xtract /home/xtract -w /home/warp/standard2diff -r /home/DTI/dti_FA -meas vol,f1samples,f2samples
This call would result in a .csv file containing the tract volume and the mean, median and standard deviation of mean_f1samples and mean_f2samples for each tract.
Usage
__ _______ ____ _ ____ _____ _ _
\ \/ /_ _| _ \ / \ / ___|_ _|__| |_ __ _| |_ ___
\ / | | | |_) | / _ \| | | |/ __| __/ _ | __/ __|
/ \ | | | _ < / ___ \ |___ | |\__ \ || (_| | |_\__ \\
/_/\_\ |_| |_| \_\/_/ \_\____| |_||___/\__\__ _|\__|___/
usage: XTRACT Stats [-h] -xtract <folder> -d <folder_basename>
[-warp <nifti> <nifti>] [-out <path>] [-meas <list>]
[-tract_list <list>] [-thr <float>]
xtract_stats: summary tract-wise measures
options:
-h, --help show this help message and exit
Required arguments:
-xtract <folder> Path to XTRACT output folder
-d <folder_basename> Path to microstructure folder and basename of data
(e.g. /home/DTI/dti_)
Optional arguments:
-warp <nifti> <nifti>
If tract are not in the same space as the data in
'-d', provide the reference image in diffusion space
(e.g. /home/DTI/dti_FA) and the xtract space to
reference space warp field
-out <path> Output filepath (Default <XTRACT_dir>/stats.csv)
-meas <list> Comma separated list of features to extract (Default =
vol,prob,length,FA,MD - assumes DTI folder has been
provided) vol = tract volume, prob = tract
probability, length = tract length Additional metrics
must follow file naming conventions. e.g. for dti_L1
use 'L1'
-tract_list <list> Comma separated list of tracts to include (default =
all found under -xtract <folder>)
-thr <float> Threshold applied to tracts (default = 0.001).
Example usage:
xtract_stats -xtract /data/xtract -d /data/Diffusion/dti_ -warp std2diff.nii.gz
XTRACT QC
Performing quality control (QC) with
xtract_qc
We provide xtract_qc for a simple way to find failed XTRACT runs and potential outliers.
The script was written by Shaun Warrington.
__ _______ ____ _ ____ _____ ___ ____
\ \/ /_ _| _ \ / \ / ___|_ _/ _ \ / ___|
\ / | | | |_) | / _ \| | | || | | | |
/ \ | | | _ < / ___ \ |___ | || |_| | |___
/_/\_\ |_| |_| \_\/_/ \_\____| |_| \__\_\\____|
usage: XTRACT QC [-h] -subject_list <txt> -out <folder> [-tract_list <list>]
[-thr <float>] [-n_std <float>] [-use_prior]
xtract_qc: quality control at the group-level
options:
-h, --help show this help message and exit
Required arguments:
-subject_list <txt> Text file containing line separated subject-wise paths
to XTRACT folders
-out <folder> Path to output folder
Optional arguments:
-tract_list <list> Comma separated list of tracts to include (default =
all found under -xtract <folder>)
-thr <float> Threshold applied to XTRACT tracts for volume
calculation (default = 0.001).
-n_std <float> The number of standard deviations (either side of mean)
to allow before being flagged as an outlier (default =
2).
-use_prior If already run xtract_qc, use previously created
metrics (default = create new metrics and overwrite)
Example usage:
xtract_qc -subject_list sublist.txt -out /data/xtract_qc
Running XTRACT QC
xtract_qc is designed to work at the group level, identifying outliers by comparison to the group mean metrics. Therefore, a cohort of xtract results are expected.
Input:
Minimally, xtract_qc requires a subject list and an output directory.
The subject list should contain N subject lines where each line is the path to a subject's XTRACT results directory.
e.g. /data/sub-001/xtract /data/sub-002/xtract /data/sub-003/xtract
xtract_qc will then search for the tract results in each subject results directory.
Output:
The primary output of xtract_qc is a html file, which should be opened using a browser. This html report contains a quick summary of the cohort and suggests potential outliers (outliers are tracts, rather than subjects) based on the recorded waytotal (i.e. number of valid streamlines generated during XTRACT) and the tract volume. The report also provides a breakdown of the number of potential outliers per tract and stores potential outliers as a .csv file.
XTRACT Divergence
Comparing connectivity blueperints with xtract_divergence
After building your connectivity blueprints with xtract_blueprint, you can use xtract_divergence to compare connectivity blueprints across individuals, groups, and species, or use xtract_divergence to translate scalar cortical maps based on the similarity between connectivity blueprints
Note
Currently xtract_divergence only supports CIFTI format connectivity blueprints. NIFTI support is coming soon!
Citations
Mars R., Sotiropoulos S.N., Passingham R.E., Sallet J., Verhagen L., Khrapitchev A.A., Sibson N., Jbabdi S. (2018) Whole brain comparative anatomy using connectivity blueprints. eLife. DOI: 10.7554/eLife.35237
Warrington S.*, Thompson E.*, Bastiani M., Dubois J., Baxter L., Slater R., Jbabdi S., Mars R.B., Sotiropoulos S.N. (2022) Concurrent mapping of brain ontogeny and phylogeny within a common space: Standardzed tractography and applications. Science Advances, 8(42). DOI: 10.1126/sciadv.abq2022
Warrington S., Bryant K., Khrapitchev A., Sallet J., Charquero-Ballester M., Douaud G., Jbabdi S.*, Mars R.*, Sotiropoulos S.N.* (2020) XTRACT - Standardised protocols for automated tractography in the human and macaque brain. NeuroImage. DOI: 10.1016/j.neuroimage.2020.116923
__ _______ ____ _ ____ _____ _ _
\ \/ /_ _| _ \ / \ / ___|_ _|_| (_)_ __
\ / | | | |_) | / _ \| | | |/ _` | \ \ / /
/ \ | | | _ < / ___ \ |___ | | (_| | |\ V /
/_/\_\ |_| |_| \_\/_/ \_\____| |_|\__,_|_| \_/
usage: xtract_divergence [-h] -bpa <CIFTI> -bpb <CIFTI> -out <folder> -masksa
<GIFTI> <GIFTI> -masksb <GIFTI> <GIFTI>
[-metric <str>]
[-roi <path,<int>,[<int>,<int>,<int>],<str>]
[-base_scalars <GIFTI> <GIFTI>]
[-target_scalars <GIFTI> <GIFTI>] [-hemi <str>]
[-names <str> <str>] [-tract_list <list>]
[-ncores <int>]
xtract_divergence: Cross-brain divergence for divergence maps and translations
options:
-h, --help show this help message and exit
Required arguments:
-bpa <CIFTI> Path to CIFTI blueprint. This is the base blueprint
-bpb <CIFTI> Path to CIFTI blueprint. This is the target blueprint:
the space in which results will be saved
-out <folder> Path to output folder (plus any filename prefixes)
-masksa <GIFTI> <GIFTI>
Path to GIFTI cortical left/right ROIs (excluding
medial wall) of the base blueprint
-masksb <GIFTI> <GIFTI>
Path to GIFTI cortical left/right ROIs (excluding
medial wall) of the target blueprint
Optional arguments:
-metric <str> Kullback-Liebler (well-behaved KLD with additional
processing, default option), KLD (no additional
processing, KLD_bias), Jenson-Shannon (JSD)
divergence, or KLD (scipy implementation, KLDscipy)
-roi <path,<int>,[<int>,<int>,<int>],<str>
ROI or greyordinate coordinates (vertex index or voxel
ijk indices) or CIFTI structure to calculate
divergence wrt whole-brain. '-roi' requires '-hemi' to
be defined.
-base_scalars <GIFTI> <GIFTI>
The scalar maps (left/right GIFTIs) to translate
between brains
-target_scalars <GIFTI> <GIFTI>
Scalar maps (left/right GIFTIs) in the target space
-hemi <str> If ROI prediction, the hemisphere of the ROI
-names <str> <str> Names of base and target brains
-tract_list <list> Comma separated tract list defining the set of tracts
to include - these must present in each brain
-ncores <int> For JSD, you may specify single-core (default) or
multi-core processing
Example call for human-to-macaque prediction of cortical MT+ ROI in the left hemisphere:
xtract_divergence -bpa human_BP.LR.dscalar.nii -bpb macaque_BP.LR.dscalar.nii
-masksa humam_atlasroi.L.shape.gii human_atlasroi.R.shape.gii
-masksb macaque_atlasroi.L.shape.gii macaque_atlasroi.R.shape.gii
-roi human_MTplus.L.shape.gii -hemi L
"-roi" flag options:
<path> to NIFTI/GIFTI binary mask. Mean blueprint in ROI used to calculate divergence to target whole-brain.
<int> single greyordinare vertex index. i.e. divergence between cortical location and whole-brain.
[<int>,<int>,<int>] ijk greyordinates. i.e. divergence between subcortical location and whole-brain.
<str> CIFTI subcortical structure. Options are those available as subcortical structures in the CIFTI convention CIFTI_STRUCTURE_* e.g. "CIFTI_STRUCTURE_THALAMUS_LEFT".
If no option supplied, will calculate dense (whole-brain to whole-brain) divergence.
Options
There are three main modes for running xtract_divergence: - whole-brain dense divergence - ROI (including single greyordinates or ROIs) to whole-brain divergence - using divergence to translate scalar maps
Whole-brain divergence
With this option, you can obtain a dense divergence matrix describing the divergence between all greyordinates in one brain to all greyordinates in another. You provide two connectivity blueprints (BPa and BPb) and their medial wall masks, e.g. call:
xtract_divergence -bpa BPa.LR.dscalar.nii -bpb BPb.LR.dscalar.nii -out ./results -masksa ./medwalla.L.shape.gii ./medwalla.R.shape.gii -masksb ./medwallb.L.shape.gii ./medwallb.R.shape.gii
The result is a CIFTI file in the same space as the second blueprint (BPb), with N volumes (where N corresponds to the number of greyordinates in BPa). Depending on the resolution of your data, this file can be very large!
ROI to whole-brain divergence
With this option, you can obtain divergence map between a given ROI in one brain to all greyordinates in another. You provide two connectivity blueprints (BPa and BPb), their medial wall masks, and some ROI (defined in the same space as BPa).
The -roi flag takes several types of input:
-
Note
You must also specify which hemisphere the ROI is in, using -hemi <L/R>.
e.g. call:
xtract_divergence -bpa BPa.LR.dscalar.nii -bpb BPb.LR.dscalar.nii -out ./results -masksa ./medwalla.L.shape.gii ./medwalla.R.shape.gii -masksb ./medwallb.L.shape.gii ./medwallb.R.shape.gii -roi AMYGDALA_LEFT -hemi L
The result will be a map showing the distribution of divergence values in the same space as BPb. Each greyordinate's value is the divergence between the greyordinate in BPb to the average connectivity fingerprint of the ROI in BPa.
Translating scalar cortical maps
With this option, you can translate a scalar cortical map (for example a myelin map, activation map, etc..) from one brain to another based on the similarity in connectivity blueprints. You provide two connectivity blueprints (BPa and BPb), their medial wall masks, and a set of base and target scalar maps.
The base maps are the maps you wish to translate to the new space. These should be in the same space as BPa. The target scalar map is simply used to ensure that the resultant prediction is saved with the correct header information. These target maps should be in the same space as BPb.
e.g. call:
xtract_divergence -bpa BPa.LR.dscalar.nii -bpb BPb.LR.dscalar.nii -out ./results -masksa ./medwalla.L.shape.gii ./medwalla.R.shape.gii -masksb ./medwallb.L.shape.gii ./medwallb.R.shape.gii -base_scalars ./myelina.L.func.gii ./myelina.R.func.gii -target_scalars ./myelinb.L.func.gii ./myelinb.R.func.gii